Role of Acetate in Fat Metabolism and Cancer Development
Cancer Development
“Cancer” refers to a wide range of conditions, but they all share one thing in common: All of them are the result of healthy cells changing into cancerous cells that multiply and spread. Medical professionals are simultaneously discovering distinct cancer risk factors to aid in cancer prevention. Despite decades of research, we still do not fully comprehend the intricate mechanism by which cancer develops. The notion that cancer is not brought on by a single event is one of the most prevalent. Multiple sources claim that normal cell changes lead to the development of malignant cells. As an internal clock, the cell cycle regulates how most healthy cells progress through various stages of life.
If enough mutations occur in the numerous genes that regulate cell growth, cells can develop cancer. Most cancer cells contained 60 or more mutations, according to research conducted by the Cancer Genome Project. It is challenging for medical professionals to ascertain which of these mutations is responsible for a specific type of cancer. This method is like trying to find a needle in a haystack because most of the mutations found in these cells have little to no effect on how cancer grows.
Metabolism of Fats in Cancer
To meet their energy requirements and produce more biomolecules, cancer cells alter their metabolism. Even within the same type of cancer, there is a significant and striking variation in the incidence of tumors. The altered metabolism of the tumor is also influenced by additional factors like diet, eating habits, exercise, and the microbiome, in addition to the genetic changes that occur independently of the cell.
Changes in lipid digestion affect the turn of events, scattering, and chemotherapy obstruction of malignant growth cells. Except for the liver and adipocytes, most adult tissues must obtain FAs and cholesterol from food. Tumors are becoming increasingly dependent on lipids from outside sources due to their ability to initiate the production of de novo fatty acids and cholesterol synthesis. This is important because it has been shown that targeting the mevalonate pathway and other enzymes related to lipogenesis can stop growth enhancement [1].
The new production of FA and cholesterol depends on fatty acid oxidation, which moves the FFAs to the mitochondria. Tumor cells also experience oxidative stress, unlike normal cells. Apoptotic pathways begin to operate because of the ROS load [2]. Malignant growth cells go through lipid digestion reinventing because these substances are fundamental for ATP union, cell division, and multiplication [3].
Acetate in Fat Metabolism
Fatty acids can be made by adipose tissue and distributed to other tissues via circulation. The oxidation process that generates energy in muscle tissue relies heavily on fatty acids as a substrate. Lipoproteins are mostly released from the liver which plays important role in lipid metabolism. Hepatocytes accumulate lipid droplets because of changes in the oxidation, production, and secretion of Very low density lipoprotein (VLDL) [4].
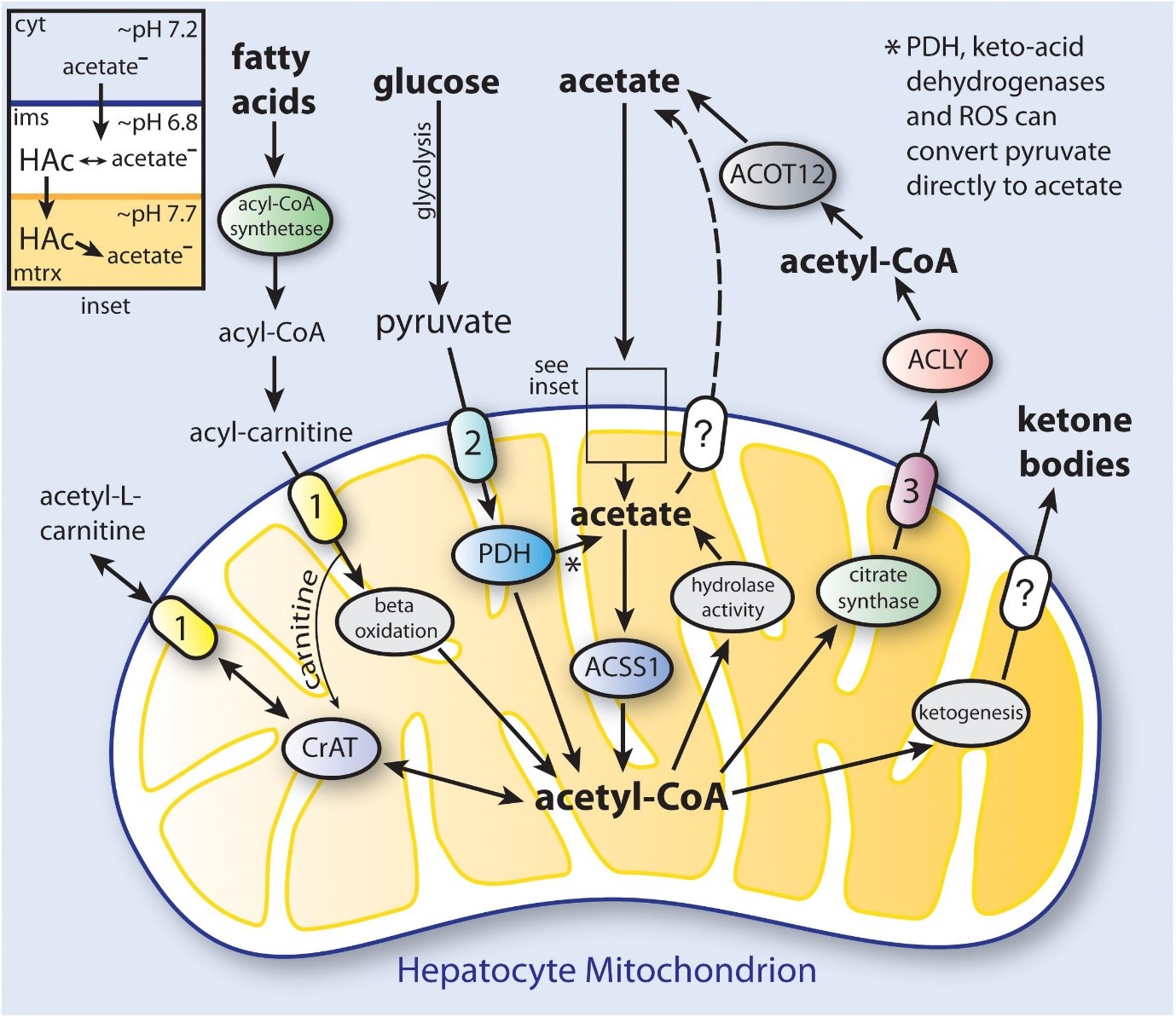
The primary byproduct of cellulose fermentation in the stomach by bacteria is acetate. Whether it comes from food or colonic fermentation, acetate has been linked to several beneficial health effects. Additionally, deacetylation processes in all cells result in the production of acetate, which is then released from several organs, including the gut and liver [5]. Cells are mostly regulated by acetate molecules. Acetate links cellular stress responses and protein function control in part through its role as a metabolic sensor.

When its intracellular levels shift, it is believed that acetyl-CoA acts as a secondary messenger, influencing a variety of metabolic processes. Acetyl-CoA, for example, is ensnared in site-explicit chromatin rebuilding since it prompts changes in histone acetylation designs when its fixation is expanded [6].
Role of Acetate in Cancer
Cancer cells usually follow Warburg effect in which oxidative phosphorylation is decreased and aerobic glycolysis is increased. The initial interpretation of these findings was that damaged mitochondria made cancer cells more dependent on enhanced glycolysis for ATP production. Two additional metabolic changes are an increase in lactate generation and higher glutaminolysis. The fact that these non-transformed cells with rapid proliferation choose to forego oxidative phosphorylation, which would produce ATP, in favor of increased pentose-phosphate shunt throughput and rapid glucose turnover, which would produce lactate, indicates that these metabolic changes are typical of these cells.
Acetate and ketone bodies may encourage the growth of cancer cells because they are straightforward carbon sources for cell development. As a result, the ketogenic diet has become more well-known as adjuvant therapy for tumors with a bad prognosis, such as glioblastoma. Alterations in mitochondrial activity are thought to be responsible for the decreased capacity of cancer cells to metabolize ketone bodies. It has been demonstrated that ACSS1 (Acyl-CoA Synthetase Short Chain Family Member 1) and ACSS2 (Acyl-CoA Synthetase Short Chain Family Member 2) are utilized by cancer cells. Some hepatocellular carcinoma cell lines, for instance, were found to prefer acetate for absorption and metabolization, while others chose glucose and used both substrates.
In SNU449 cancer cells, knocking down ACSS2 and ACSS1 decreased acetate absorption and cell survival, favouring acetate significantly over glucose. When oxygen and nutrients are scarce, there is increasing evidence that ACSS2 uses transcription factor complexes to switch from regulating lipogenesis to regulating metabolic and signaling processes. Under normal conditions, lipid production and biomass increase in cancer cells because glycolysis and glutaminolysis provide the main carbon sources. Acetate is a crucial alternate source of carbon for cancer cells. Acetate, glucose, and amino acids like glutamine and glutamate are the top three sources of acetyl-CoA in cancer cells.
The labelling of fatty acids and lipids under hypoxia and low serum circumstances was linked to the change toward acetate consumption in cancer cells, according to research, and both of these processes need ACSS2. Cancer cells can also multiply under stress conditions. Additional stresses include a lack of nutrients and extracellular acidification caused by processes like glycolysis and elevated lactic acid production. Most cancer cells do not make significant use of ketone bodies if glucose is available. Instead of being defective as Warburg proposed, cancer cells are now believed to be extremely dependent on the excess of glutamate and glucose. Ketone body and acetate consumption are not necessary for cells to activate in response to a nutritional deficiency when they primarily use glucose for aerobic glycolysis.
In vitro, acetate has also been shown to kill thymic tumor cells at high doses, but not healthy thymocytes at the same concentration (12.5 mM). Acetate supplementation may be used as adjuvant therapy in the treatment of some cancers, although it is not significant in normal physiology. It is believed that distinct pathways for the cytotoxicity of acetate exist in several types of cancer cells. These might involve an increase in acetate-induced lysosomal and mitochondrial permeabilization. It is also known that acetate kills tumor cells by activating caspases. Cell-specific effects can frequently result from acetate-based epigenetic processes that regulate gene transcription. Acetylation inhibits ACSS2 function, so it makes sense that extremely high concentrations of it decrease its activity.
All ACSS enzymes have been linked to the cancer spreading. The oxidation of acetic acid derivation and propionate by the mitochondrial structures ACSS1 and ACSS3 in all likelihood contributes fundamentally to energy creation, but they may not contribute as a lot to administrative exercises in malignant growth cells as ACSS2. We do not know of any studies that have looked into the possibility that ACSS1 aids certain acetylation processes in the mitochondrial.

References
- Sadowski, M. C., Pouwer, R. H., Gunter, J. H., Lubik, A. A., Quinn, R. J., & Nelson, C. C. (2014). The fatty acid synthase inhibitor triclosan: repurposing an anti-microbial agent for targeting prostate cancer. Oncotarget, 5(19), 9362.
Link: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4253440/
- Horiguchi, Akio, et al. “Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells.” The Journal of urology 180.2 (2008): 729-736.
Link: https://www.auajournals.org/doi/abs/10.1016/j.juro.2008.03.186
- Lee, H. R., Hwang, K. A., Nam, K. H., Kim, H. C., & Choi, K. C. (2014). Progression of breast cancer cells was enhanced by endocrine-disrupting chemicals, triclosan and octylphenol, via an estrogen receptor-dependent signalling pathway in cellular and mouse xenograft models. Chemical research in toxicology, 27(5), 834-842.
Link: https://www.auajournals.org/doi/abs/10.1016/j.juro.2008.03.186
- Grabacka, M., Pierzchalska, M., Dean, M., & Reiss, K. (2016). Regulation of ketone body metabolism and the role of PPARα. International journal of molecular sciences, 17(12), 2093.
Link: https://www.mdpi.com/1422-0067/17/12/2093
- Moschen, I., Bröer, A., Galić, S., Lang, F., & Bröer, S. (2012). Significance of short-chain fatty acid transport by members of the monocarboxylate transporter family (MCT). Neurochemical research, 37(11), 2562-2568.
Link: https://link.springer.com/article/10.1007/s11064-012-0857-3
- Liu L, Fu C, Li F. Acetate Affects the Process of Lipid Metabolism in Rabbit Liver, Skeletal Muscle and Adipose Tissue. Animals (Basel). 2019 Oct 14;9(10):799. doi: 10.3390/ani9100799. PMID: 31615062; PMCID: PMC6826666.
Link: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6826666/#:~:text=Acetate%20decreased%20the%20lipid%20synthesis,acetate%2Dreduced%20intracellular%20fat%20content.
- Arun, P., Madhavarao, C. N., Moffett, J. R., Hamilton, K., Grunberg, N. E., Ariyannur, P. S., … & Namboodiri, A. (2010). Metabolic acetate therapy improves phenotype in the tremor rat model of Canavan disease. Journal of inherited metabolic disease, 33(3), 195-210.
Link: https://link.springer.com/article/10.1007/s10545-010-9100-z
- Arun, P., Madhavarao, C. N., Moffett, J. R., & Namboodiri, M. A. (2006). Regulation of N‐acetylaspartate and N‐acetylaspartylglutamate biosynthesis by protein kinase activators. Journal of neurochemistry, 98(6), 2034-2042.
Link: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1471-4159.2006.04068.x
- Arun, P., Madhavarao, C. N., Moffett, J. R., & Namboodiri, A. M. (2008). Antipsychotic drugs increase N‐acetylaspartate and N‐acetylaspartylglutamate in SH‐SY5Y human neuroblastoma cells. Journal of neurochemistry, 106(4), 1669-1680.
Link: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1471-4159.2008.05524.x
- Gao, X., Lin, S. H., Ren, F., Li, J. T., Chen, J. J., Yao, C. B., … & Lei, Q. Y. (2016). Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun 7: 11960.
Link: https://www.diva-portal.org/smash/record.jsf?pid=diva2%3A523865&dswid=6367
- Hagenfeldt, L., Bollgren, I., & Venizelos, N. (1987). N‐acetylaspartic aciduria due to aspartoacylase deficiency—a new aetiology of childhood leukodystrophy. Journal of inherited metabolic disease, 10(2), 135-141.
Link: https://onlinelibrary.wiley.com/doi/abs/10.1007/BF01800038
- Schug, Z. T., Peck, B., Jones, D. T., Zhang, Q., Grosskurth, S., Alam, I. S., … & Gottlieb, E. (2015). Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer cell, 27(1), 57-71.
Link: https://www.sciencedirect.com/science/article/pii/S153561081400511X